



















Single Cell RNA Sequencing (scRNA-Seq)
- Data Processing Overview
- Bulk RNA Sequencing (RNA-seq)
- Single Cell RNA Sequencing (scRNA-seq)
- Amplicon Sequencing
- Metagenomics
- Platform-Specific Microarrays
- Methylation Sequencing (Methyl-seq)
Introduction
Single-cell RNA sequencing (scRNA-Seq) is a powerful method that measures gene expression in individual cells, rather than averaging across a whole tissue or organism. In this process, RNA is isolated from each cell, converted into cDNA, and labeled with unique barcodes before sequencing. This allows researchers to identify which genes are expressed in specific cells, making it possible to distinguish different cell types (e.g., healthy vs. cancer cells) or track cells at various stages of development. Because of this fine resolution, scRNA-Seq provides detailed insights into the diversity and function of cells within complex biological samples.
Unlike bulk RNA sequencing, which combines RNA from all cells into a single measurement, scRNA-Seq preserves the individuality of each cell. Bulk RNA-Seq can be thought of as a fruit smoothie—where all the fruit is blended together indiscriminately—while scRNA-Seq is more like a fruit salad, where individual fruit pieces can be picked out and identified. This single-cell approach enables researchers to detect rare cell types, study cellular heterogeneity, and better understand processes like development, disease progression, and drug responses. Its ability to capture these differences makes scRNA-Seq a transformative tool in fields such as molecular biology, medicine, and drug discovery.
GeneLab Sample Processing Standard Operating Procedures
GeneLab offers publicly available Standard Operating Procedures (SOPs) for sequencing prepared libraries. These protocols can be accessed and implemented by researchers in their own institutions to ensure consistency and reproducibility. Explore and download the GeneLab SOPs on GitHub.
GeneLab Data Processing Capabilities
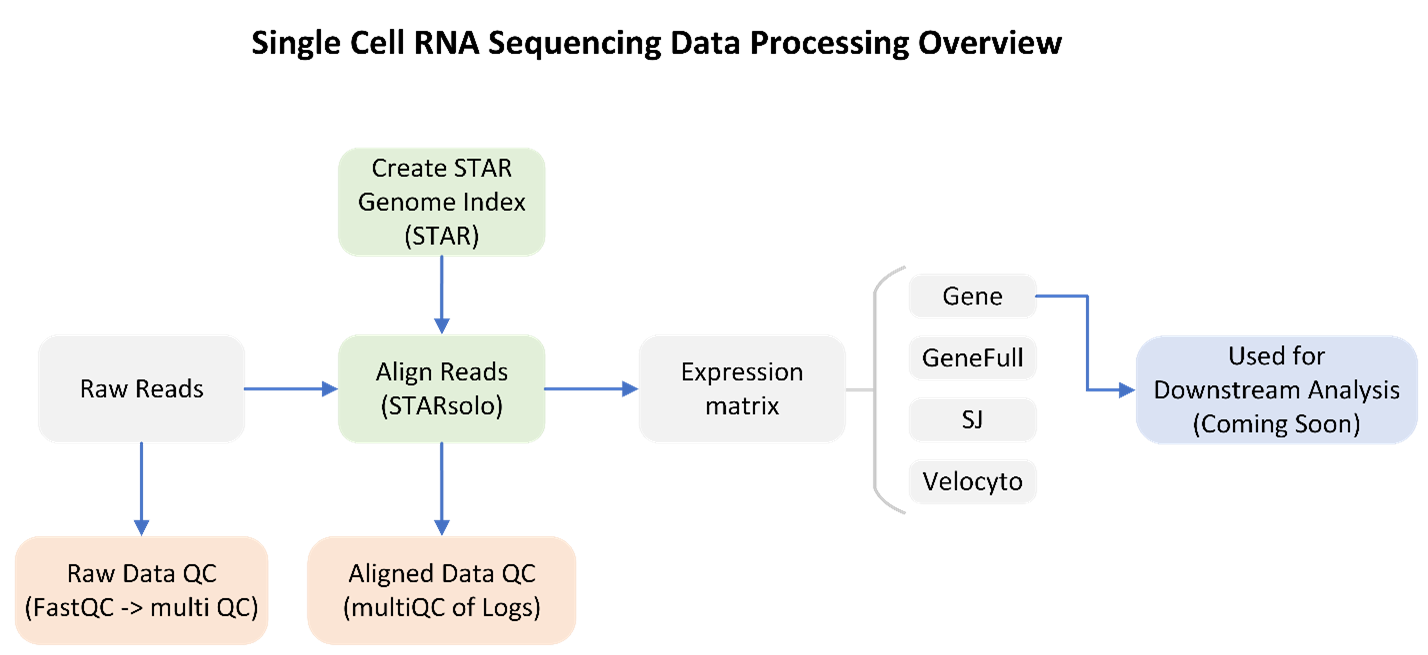
Consensus Processing Pipeline
GeneLab worked with the scientific community via the Analysis Working Groups (AWGs) to develop a consensus pipeline for processing scRNA-Seq data hosted on the Open Science Data Repository (OSDR), to generate raw gene counts per cell for each sample. The raw gene counts can then be used in downstream analyses to identify spaceflight-induced differentially expressed genes in individual cell types. Each step of the pipeline and the respective output files generated are publicly available on the scRNA-Seq page of the GeneLab Data Processing GitHub repository.
Data Processing Workflow
The GeneLab Data Processing Team is currently working on wrapping the scRNA-Seq pipeline into a Nextflow workflow. This workflow will be used to process all scRNA-Seq datasets hosted on OSDR, and the GeneLab processed data products will be made publicly available alongside each dataset on OSDR. The workflow will be made publicly available on GitHub, along with instructions for how to install and run the workflow to allow users to re-process OSDR data or process their own scRNA-Seq data using the GeneLab workflow.